Novel Subgenotypes of Bovine Viral Diarrhea Virus based on 5’ UTR Molecular Epidemiology in Cattle from Huhhot of Inner Mongolia Autonomous Region, China.
Feng-Xue Wanga, Wudong Gaowaa, Chunyu Liua, Junhao Fana, Kun Yina, b, Yiming Li, Yuming Suna, Bingwu Zhengc, Yang Yangd, Hongzhe Zhaoa, Hao Wanga, Xinpei Lia, Zipeng Maa, Ming Jina, Qianjin Songa, He Zhanga, Yong-Jun Wena
A. College of Veterinary Medicine, Key laboratory for clinical diagnosis and treatment of animal diseases of ministry of agriculture, Inner Mongolia Agricultural University, Inner Mongolia Autonomous Region, China
B. State Key Laboratory for Molecular Biology of Special Economic Animals, Institute of Special Economic Animals and Plants, Chinese Academy of Agricultural Sciences CAAS, Changchun, Jilin 130112, China.
C. Management office, Hohhot city zoo.
D. The State Key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock, Inner Mongolia University.
Corresponding author:
Yong-Jun Wen, College of Veterinary Medicine, Inner Mongolia Agricultural University, People’s Republic of China.
Received: October 2, 2021
Accepted: October 13, 2021
Published: October 15, 2021
1. Abstract
1.1.Bovine viral diarrhea (BVD) causes high economic losses in the cattle population worldwide. Here, we present the results of an epidemiological survey for Bovine viral diarrhea virus (BVDV) in Northwest of China. In this study, a total of 167 samples were collected from the bovine farms and cattle slaughters for epidemiology nearby Huhhot in Inner Mongolia Autonomous Region during 2017 and 2018. Positive BVDV isolates were genotyped based on a comparison of gene sequences from their 5’untranslated regions (5’UTR). Results indicated that, out of 167 samples, 68 (40.72%) were BVDV-RNA-positive. 28 of them were sequenced and analyzed with 5’UTR and used for constructing phylogenetic tree. Phylogenetic analysis based on 5’UTR revealed that13 of the BVDV isolates belong to BVDV-1 and 15 belong to BVDV-2 genotype. Interesting, two novel BVDV-1 subgenotypes and one novel BVDV-2 subgenotype were found in present study. Therefore, the result of this study will be useful to understand epidemiology in Inner Mongolia Autonomous Region and allow producers to better protect their livestock.
1.2.Keywords: Bovine viral diarrhea viruses, Epidemiology, 5’UTR, Novel Genotype.
Importance:
An epidemiological survey indicated two novel BVDV-1 subgenotypes and one novel BVDV-2 subgenotypewere found based on 5’UTR phylogenetic tree in Northwest of China.
Introduction
BVDV can cause bovine respiratory, gastroenteric and reproductive clinical consequences, and still lead to the birth of immunotolerant. It is the main viral infectious disease of cattle, which is a worldwide epidemic disease. Bovine viral diarrhea virus (BVDV) is a member of the genus Pestivirus. According to the genetic characteristics of the genome, BVDV is divided into two genotypes, BVDV-1 and BVDV-2. At present, BVDV-1 strains have further been divided into 21 subtypes (1a-1u) (Tajima et al., 2001; Vilcek et al., 2001; Yamamoto et al., 2008; Gong et al., 2013; Deng M. et al., 2015). To date, the BVDV circulating in the Chinese cattle population is mainly BVDV-1b, -1c, -1m, -1p (Zhong et al., 2011). According to the difference of secondary structure of 5’ -UTR sequence, BVDV-2 is divided into 4 subtypes, BVDV-2 a, 2 b, 2 c and 2 d (Giangaspero et al., 2008).
Bovine viral diarrhea virus infection in pregnant animals can also result in the birth of a persistently infected (PI) calf. PI animals are the main source of virus transmission to susceptible animals. It is an obstacle for the elimination of the virus in a susceptible population. The combined economic impact of BVDV has been estimated at a 20 to 57 million dollar loss per million calving’s in the USA. It also caused a huge economic loss from BVDV in China. 5’-UTR, Npro, and E2 genes were also used for genetic typing of the pestiviruses (Vilcek et al., 2001; Vijayaraghavan et al., 2012).
Variability of BVDV is distinct. A growing number of BVDV-1 and BVDV-2 subgenotypes based on phylogenetic analysis indicated the BVDV is genetically highly heterogeneous. The highest number of various BVDV subgenotypes has been documented in European countries (Yesilbag et al., 2017). Different genomic regions, i.e., 5’UTR (Beer et al., 2002; Becher et al., 2003; O’Brien et al., 2017), Npro (Maya et al., 2016; O’Brien et al., 2017), E2 (Couvreur et al., 2002; Yilmaz et al., 2012), have been used for genotyping and classification of BVDV. Partial 5’UTR sequences have been most frequently used for phylogenetic analyses and genotyping of BVDV isolates. A 288bp size of 5’UTR was used for epidemiology of BVDV in present study.
The epidemiology and genetic variations analysis of BVDV can implicate in disease control as diagnostics and vaccines that work well against homologous strains can be less efficacious for genetically- distinct viruses.
Materials and methods
Sample collection and processing
The 167 whole blood, lung tissue and intestinal contents samples were collected from the cattle of six bovine slaughters and calf and bovine of 26 farms, which were healthy or showed minor respiratory disease, in Huhhot and its surrounding rural breeding area of Inner Mongolia Autonomous Region, China during 2017 and 2018. Blood samples were collected for diagnostic purposes by venipuncture into silicon-coated vacutainer tubes and were immediately transported to the laboratory and stored at -20OC in tubes appropriate for freezing. Other tissue samples were homogenized in 2 ml of PBS and centrifuged at a 2,000×g for 3 min to remove the suspended solids. The supernatants were stored at −80OC until testing.
RNA extraction and RT-PCR
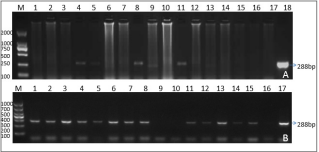
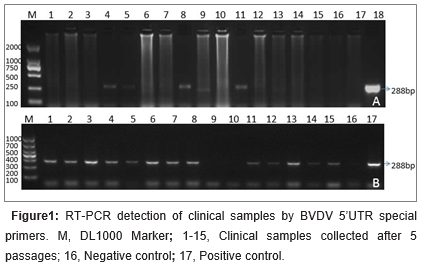
Total RNA was extracted from whole blood or tissues using the TRIzol reagent (Life Technologies, U.S.) according to the manufacturer’s instructions. Viral cDNA was constructed using reverse transcriptase (M-MLV, Invitrogen) with 10 μL of RNA. The polymerase chain reaction (PCR) amplification was performed with 5 μL of cDNA as the template using previous detection primers BVP1 (5’TAGCCATGCCCTTAGTAGGAC-3’) and BVP2 (5’CTCCATGTGCCATGTACAGCA-3’) (Vilcek et al., 2003) flank a 288-bp DNA fragment for BVDV 5’UTR with DNA polymerase Pfu (NEB, US). The PCR amplification was performed for 35 cycles: denaturation at 94°C for 30 s, annealed at 57°C for 30 s, and elongation at 72°C for 30 s. The samples were then incubated for an additional 10 min at 72°C and cooled to 4°C until further processing. Five microliters of the PCR products were analyzed on agarose gel (1.5%) electrophoresis at 120 V for 20 min.
Sequencing and Phylogenetic analysis of nucleotides
Twenty-eight isolates were randomly chosen from RNA positive samples for sequencing within the 5’UTR of the genome. The amplicons were purified using an Omega gel extraction kit (Omega, U.S.) and sequenced. These nucleotide sequences were assembled and proof read used the SeqMan program of Lasergene package (DNASTAR Inc., USA). For phylogenetic analysis, a total of 79 related reference sequences of 5’UTR (Listed in Table 1) were retrieved from the NCBI GenBank database (http://www.ncbi.nlm.nih.gov/genbank) and field strains in present study were also included for comparison (28 strains). Some of reference strains are reported as classified subgenotypes. A phylogenetic tree was constructed by the UPGMA method with MEGA7.0. The evolutionary distances were computed using the Maximum Composite Likelihood method. The GenBank accession numbers for the sequences of 5’UTR genes of the 28 BVDV isolates in present paper are listed in Table 1
Table 1: BVDV isolates and reference strains described in this study
Notes: The bold letters are strains isolated in present study.
Results
RT-PCR survey of clinical samples
RT-PCR assays were performed to determine the BVDV species with 5’UTR specific primers responsible for the infection. Of the 167 samples, 68 (40.72%) were BVDV-RNA-positive by RT-PCR. Of the products 28 were sequenced and blasted in NCBI GenBank. Thirteen BVDV-1 isolates (47.83%) were genotyped. Fifteen (52.17%) were classified as BVDV-2 in all the sequenced28 antigen positive samples. The electrophoresis results of RT-PCR products of some samples were shown in figure 1a. A summary of the isolates and reference strains is presented in Table 1. The 5’UTR genes of these isolates were used for further phylogenetic analysis.
Subgenotypes based on phylogenetic analysis of 5’UTR of BVDV-1 in Inner Mongolia
1. The nucleotide sequences of field viruses and a number of known reference strains representative of all known species and subtypes of BVDV were aligned and phylogenetic trees were constructed. It displayed phylogenetic trees for a selection of the 5’UTR in Fig.
2. The specified fragments of 5’UTR region (288bp) from cDNA preparations were detected and sequenced. Phylogenetic analysis was performed based on the 288bp fragments of 13 BVDV-1 strains of 68 BVDV-positive samples collected from cattle’s in Inner Mongolia between 2017 and 2018. Thirty-six B reference strains from GenBank were used. BVDV-1 isolates analyzed in this work were deposited in GenBank under following accession numbers: MK204893-MK204905 (Listed in Table 1). The topology of the tree (Figure 2a) showed that all 49 BVDV-1 strains belonged to 20 distinct subgenotypes, namely BVDV-1a (n=2),BVDV-1b (n=4), BVDV-1c (n=8), BVDV-1d (n=4), BVDV-1e (n=2), BVDV-1f (n=2), BVDV-1g (n=4), BVDV-1h (n=2), BVDV-1i (n=2), BVDV-1j (n=3), BVDV-1k (n=1), BVDV-1l (n=1), BVDV-1m (n=4), BVDV-1n (n=2), BVDV-1o (n=1), BVDV-1p (n=2), BVDV-1q (n=1), BVDV-1r (n=2), BVDV-1s(n=1), BVDV-1t(n=1), BVDV-1u(n=1), BVDV-1v(n=2), BVDV-1w(n=2). The subgenotypes BVDV-1m, -1n, -1o, -1p, and -1q had been detected exclusively in Asia. Similarly, BVDV -1f, -1g,-1h, -1k, -1l, -1r, 1s, and -1t have not been reported to occur in countries outside Europe(Giammarioli et al., 2008; Yilmaz et al., 2012; Factor et al., 2016; Gomez-Romero et al., 2017; Silveira et al., 2017; Yesilbag et al., 2017). However, phylogenetic analysis clustered the 13 BVDV-1 isolates into six subgenotypes in present study: BVDV-1a, BVDV-1c, BVDV-1d, BVDV-1m, and two potentially novel subgenotypes, tentatively designated as ‘BVDV-1v’ and ‘BVDV-1w’ in present study (Figure 2).
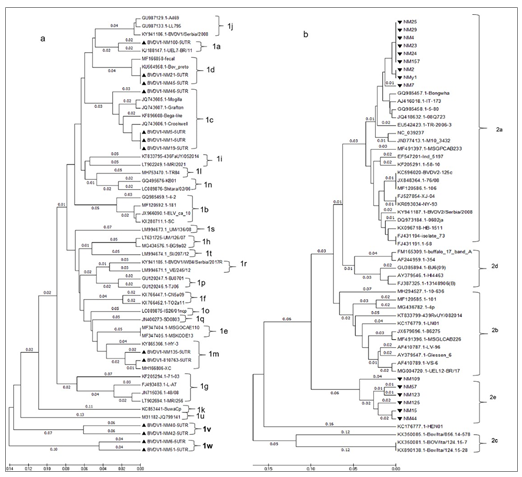
UEL-BR/11 isolated from Brazil, which was belong to BVDV-1a. There are 4 strains: NM1, NM5, NM19 and NM46 in the BVDV-1c group, which also including 4 strains from Australia. The NM21 and NM45 were classified as BVDV-1d. ELV_ca_10 strain from Denmark belonging to BVDV-1d in a previous work was not clustered into BVDV-1d but BVDV-1b in present work. 810763 and NM135 strains isolated from bovine herd located west of Inner Mongolia were classified as BVDV-1m. The first BVDV-1m strain, ZM-95, was initially isolated from swine herds in Inner Mongolia in 1995(Wang X, 1996). Six BVDV-1m isolates from Beijing were detected in 2015(Weng et al., 2015). Accumulating evidence is indicating that BVDV-1m also is a predominant subgenotype in cattle herd (Deng Y. et al., 2012; Gao et al., 2013).
Interesting, the other four strains NM6, NM51, NM40 and NM42 were shown clustering into two novel distinct phylogenetic groups from BVDV-1a~1u. Here, we first tentatively named them as ‘BVDV-1v’ and ‘BVDV-1w’. When NM6 is blasted with other strains in GenBank, the homology is up to 90%. There is highest identity between NM6 and MF-2, a BVDV-1c strain from China. Though NM51 is belong to same cluster with NM6, it has an 84% identity with BJ09_21 strain, which is also from China. NM40 only has a homology up to 82% with other strains in GenBank, which are belong to BVDV-1d and BVDV-1a when blasting. NM40 is likely origin from has most similar with AL3a@10, a BVDV-1d isolate from Italy. We deduced NM40 are likely origin from recombination with BVDV-1a and BVDV-1d.
Subgenotypes based on phylogenetic analysis of 5’UTR of BVDV-2 in Inner Mongolia
The 15 positive samples belonged to BVDV2, which were divided into 2 different subgenotypes (Figure 2b). Phylogenetic analysis results showed that 60% (9/15) of the sequences (NM2, NM4, NM7, NM23, NM24, NM25, NM29, NM157, NMy1) were typed into a single subgroup BVDV-2a. Specially, other 6 strains (NM15, NM44, NM57, NM109, NM123, NM125) were characterized as a new subgroup that is different from BVDV-2a~BVDV-2d. These six isolates could not be assigned to any known BVDV-2 subgenotype. We appointed it as ‘BVDV-2e’. Subgenotypes BVDV-2b, -2c, and -2d strains, which have been reported in South America, were not detected in current study. The BVDV-2a subgenotype appears to be more common than the other subgenotypes worldwide.
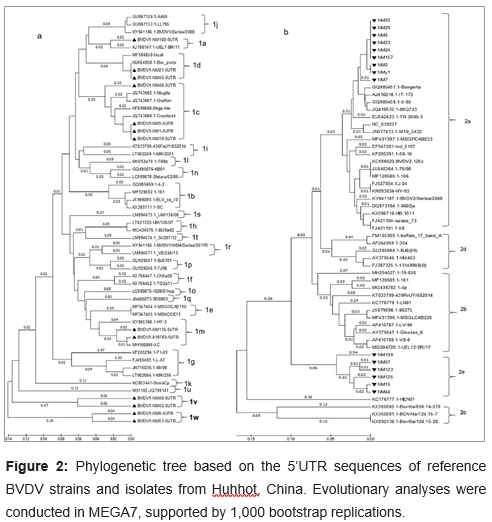
a. Phylogenetic tree showing the genetic relationship between Bovine viral diarrhea virus 1 isolates based on analysis of 49 nucleotides derived from the 5’UTR. The tree contains 13 BVDV-1 isolates sequenced in this study plus 36 reference strains (shown in Table 2). The studied strains were labeled with a symbol
b. The phylogenetic tree was generated based on comparison of nucleotide sequences of the 5’UTR of 15 BVDV-2 isolates with 39 reference sequences (shown in Table 2) downloaded from the GenBank database. The 15 BVDV-2 isolates are indicated in symbol Numbers at the phylogenetic branches indicate branch lengths (next to the branches) in the same units. The sequences for reference strains were listed with strain name and GenBank no.
Discussion
Were detected more than other 4 groups 1a, 1m, 1v and 1w.
Deng et al. investigated BVDV genotypes in four bovine species to found BVDV-1b, BVDV-1m and a new cluster BVDV-1u were dominant subtypes in China (Deng M. et al., 2015). However, they collected the samples from east but not west of Inner Mongolia. We mainly located in west of Inner Mongolia to supervise the prevalence of BVDV. Importantly, we found not only the subgenotypes BVDV- 1m but also two novel clusters BVDV-1v and BVDV1w. It indicated that more complicated BVDV strains were spread in west of Inner Mongolia.
Infection with BVDV-2 was first described in North America in the early of 1990s (Corapi et al., 1989). BVDV-2 was found in cattle in Xinjiang Autonomous Region and in Qinghai province of China (Gong et al., 2014). The BVDV-2a subgenotype appears to be more common than the other subgenotypes worldwide (Giammarioli et al., 2008; Oguzoglu et al., 2010; Behera et al., 2011; Han et al., 2016; Gomez-Romero et al., 2017). A recent phylogenetic analysis of BVDV in Mongolia revealed BVDV-1a and BVDV-2a were dominant genotype (Ochirkhuu et al., 2016). Though Inner Mongolia of China locates nearby Mongolia, the BVDV genotype based on epidemiological studies showed more subgenotypes appeared in Inner Mongolia of China. Two BVDV-2 subgenotypes were found in present study.
Here, we present the results of an epidemiological survey for Bovine viral diarrhea virus (BVDV) in Inner Mongolia, China, especially in Huhhot and surrounding areas of Inner Mongolia Autonomous Region. Our investigation describes the genetic diversity of BVDV from cattle in Huhhot of Inner Mongolia. It provides important information for the clinician and diagnostician responsible for diagnosis of BVDV.
To summarize, the presence of novel subgenotypes BVDV-1v and BVDV-1w and BVDV-2e were described in China dairy cattle for the first time. Further studies are required to investigate the prevalence of BVDV infection in a larger cattle population as well as the role in various clinical conditions and economical losses. The presence of new species is important for the evaluation of current diagnostic protocols and for the development of control programs, such as vaccination. Furthermore, it underlines the necessity to quarantine and test imported cattle thoroughly before introduction into local herds.
Acknowledgements
This work has been funded in whole or in part with Introducing Talents Scientific Research Project of Inner Mongolia Agricultural University ‘Research on prevention and control of herbivore animal diseases and biological products’ (NDGCC2016-22 and NDYB2018-
and National Natural Science Foundation of China (NSFC) ‘Molecular mechanism for synaptic recycling dysfunction induced by G protein of street rabies virus in primary mouse neural cells’ (31572505). The study was also supported with Key Laboratory of Clinical Diagnosis and Treatment Technology in Animal Disease, Ministry of Agriculture, P.R China and Ruminant Animal Disease Diagnosis center, Inner Mongolia Agricultural University.
Author contributions:
FXW, YJW contributed to the design of the work; JHF, WDGW, KY, XPL, ZPM, MJ HZ, and YY performed the experiments in the study. FXW, YML, YMS, HW, CYL, BWZ, HZZ, and QJS analyzed the data. FXW, YJW wrote the manuscript. All authors read and approved the final manuscript.
References:
1. Becher P, Avalos Ramirez R, Orlich M, Cedillo Rosales S, Konig M, Schweizer M,et.al. Genetic and antigenic characterization of novel pestivirus genotypes: implications for classification. Virology. 2003; 311: 96-104.
2. Beer M, Wolf G, Kaaden OR. Phylogenetic analysis of the 5’-untranslated region of german BVDV type II isolates. J Vet Med B Infect Dis Vet Public Health. 2002; 49: 43-47.
3. Behera SP, Mishra N, Vilcek S, Rajukumar K, Nema RK, Prakash A, et.al. Genetic and antigenic characterization of bovine viral diarrhoea virus type 2 isolated from cattle in India. Comp Immunol Microbiol Infect Dis. 2011; 34: 189-196.
4. Corapi WV, French TW, Dubovi EJ. Severe thrombocytopenia in young calves experimentally infected with noncytopathic bovine viral diarrhea virus. J Virol. 1989; 63: 3934-3943.
5. Couvreur B, Letellier C, Collard A, Quenon P, Dehan P, Hamers C, et.al. Genetic and antigenic variability in bovine viral diarrhea virus (BVDV) isolates from Belgium. Virus Res.2002; 85: 17-28.
6. Deng M, Ji S, Fei W, Raza S, He C, Chen Y, et.al. Prevalence study and genetic typing of bovine viral diarrhea virus (BVDV) in four bovine species in China. PLoS One. 2015; 10: e0121718.
7. Deng Y, Sun CQ, Cao SJ, Lin T, Yuan SS, Zhang HB, et.al. High prevalence of bovine viral diarrhea virus 1 in Chinese swine herds. Vet Microbiol. 2012; 159: 490-493.
8. Factor C, Yus E, Eiras C, Sanjuan ML, Cervino M, Arnaiz I and et.al. Genetic diversity of bovine viral diarrhea viruses from the Galicia region of Spain. Vet Rec Open. 2016; 3: e000196.
9. Gao S, Luo J, Du J, Lang Y, Cong G, Shao J,et.al. Serological and molecular evidence for natural infection of Bactrian camels with multiple subgenotypes of bovine viral diarrhea virus in Western China. Vet Microbiol. 2013; 163: 172-176.
10. Giammarioli M, Pellegrini C, Casciari C, Rossi E, De Mia GM. Genetic diversity of bovine viral diarrhea virus 1: Italian isolates clustered in at least seven subgenotypes. J Vet Diagn Invest. 2008; 20: 783-788.
11. Giangaspero M, Harasawa R, Weber L, Belloli A. Genoepidemiological evaluation of Bovine viral diarrhea virus 2 species based on secondary structures in the 5’ untranslated region. J Vet Med Sci, 2008; 70: 571-580.
12. Gomez-Romero N, Basurto-Alcantara FJ, Verdugo-Rodriguez A, Bauermann FV, Ridpath JF. Genetic diversity of bovine viral diarrhea virus in cattle from Mexico. J Vet Diagn Invest. 2017; 29: 362-365.
13. Gong X, Cao X, Zheng F, Chen Q, Zhou J, Yin H, et.al. Identification and characterization of a novel subgenotype of bovine viral diarrhea virus isolated from dairy cattle in Northwestern China. Virus Genes. 2013; 46: 375-376.
14. Gong X, Liu L, Zheng F, Chen Q, Li Z, Cao X, et.al. Molecular investigation of bovine viral diarrhea virus infection in yaks (Bos gruniens) from Qinghai, China. Virol J. 2014; 11: 29.
15. Han YJ, Chae JB, Chae JS, Yu DH, Park J, Park BK. Identification of bovine viral diarrhea virus infection in Saanen goats in the Republic of Korea. Trop Anim Health Prod. 2016; 48: 1079-1082.
16. Maya L, Puentes R, Reolon E, Acuna P, Riet F, Rivero R. Molecular diversity of bovine viral diarrhea virus in uruguay. Arch Virol. 2016; 161: 529-535.
17. O’Brien E, Garvey M, Walsh C, Arkins S, Cullinane A. Genetic typing of bovine viral diarrhoea virus in cattle on Irish farms. Res Vet Sci. 2017; 111: 14-20.
18. Ochirkhuu N, Konnai S, Odbileg R, Odzaya B, Gansukh S, Murata S, et.al. Molecular detection and characterization of bovine viral diarrhea virus in Mongolian cattle and yaks. Arch Virol. 2016; 161: 2279-2283.
19. Oguzoglu TC, Muz D, Yilmaz V, Alkan F, Akca Y, Burgu I. Molecular characterization of Bovine virus diarrhea viruses species 2 (BVDV-2) from cattle in Turkey. Trop Anim Health Prod. 2010; 42: 1175-1180.
20. Silveira S, Weber MN, Mosena AC, da Silva MS, Streck AF, Pescador CA, et.al. Genetic Diversity of Brazilian Bovine Pestiviruses Detected Between 1995 and 2014. Transbound Emerg Dis. 2017; 64: 613-623.
21. Tajima M, Frey HR, Yamato O, Maede Y, Moennig V, Scholz H, et.al. Prevalence of genotypes 1 and 2 of bovine viral diarrhea virus in Lower Saxony, Germany. Virus Res. 2001; 76: 31-42.
22. Vijayaraghavan B, Xia H, Harimoorthy R, Liu L, Belak S. Evaluation of envelope glycoprotein E (rns) of an atypical bovine pestivirus as antigen in a microsphere immunoassay for the detection of antibodies against bovine viral diarrhea virus 1 and atypical bovine pestivirus. J Virol Methods. 2012; 185: 193-198.
23. Vilcek S, Greiser-Wilke I, Durkovic B, Obritzhauser W, Deutz A, Kofer J. Genetic diversity of recent bovine viral diarrhoea viruses from the southeast of Austria (Styria). Vet Microbiol. 2003; 91: 285-291.
24. Vilcek S, Paton DJ, Durkovic B, Strojny L, Ibata G, Moussa A, et.al. Bovine viral diarrhoea virus genotype 1 can be separated into at least eleven genetic groups. Arch Virol. 2001; 146: 99-115.
25. Wang X TC, Li H, Jin K, Xuan H, Chang G, Sun H, et.al. Pigs naturally infected by bovine diarrhea virus present signs resembling hog cholera. Chin J Vet Sci. 1996; 341-345.
26. Weng XG, Song QJ, Wu Q, Liu MC, Wang ML, Wang JF. Genetic characterization of bovine viral diarrhea virus strains in Beijing, China and innate immune responses of peripheral blood mononuclear cells in persistently infected dairy cattle. J Vet Sci. 2015; 16: 491-500.
27. Xue F, Zhu YM, Li J, Zhu LC, Ren XG, Feng JK, et.al. Genotyping of bovine viral diarrhea viruses from cattle in China between 2005 and 2008. Vet Microbiol. 2010; 143: 379-383.
28. Yamamoto T, Kozasa T, Aoki H, Sekiguchi H, Morino S, et.al. Genomic analyses of bovine viral diarrhea viruses isolated from cattle imported into Japan between 1991 and 2005. Vet Microbiol. 2008; 127: 386-391.
29. Yesilbag K, Alpay G, Becher P. Variability and Global Distribution of Subgenotypes of Bovine Viral Diarrhea Virus. Viruses. 2017; 9.
30. Yilmaz H, Altan E, Ridpath J, Turan N. Genetic diversity and frequency of bovine viral diarrhea virus (BVDV) detected in cattle in Turkey. Comp Immunol Microbiol Infect Dis. 2012; 35: 411-416.
31. Zhong F, Li N, Huang X, Guo Y, Chen H, Wang X, et.al. Genetic typing and epidemiologic observation of bovine viral diarrhea virus in Western China. Virus Genes. 2011; 42: 204-207.